Research
The use of computers to study a wide range of chemical and biological phenomena has increased with increasing computational power. Simulation of biological systems using classical and combined quantum/classical methods has provided insights into a variety of biological processes. In the Cisneros group, we are developing new methods for biosimulations and employing computational simulations to study biomolecular systems. The current research directions in our group are:
Computational investigation of DNA modification enzymes
DNA repair and replication is carried out by DNA polymerases. These enzymes are involved in the maintenance of genome stability which is critical for the survival of DNA-based organisms. Errors in either of these two processes can result in mutations, some of which can lead to disease or even death. The understanding of the mechanisms carried out by these proteins at the atomic level could provide insights into these processes.
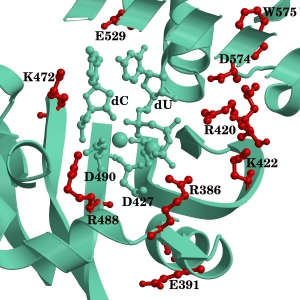
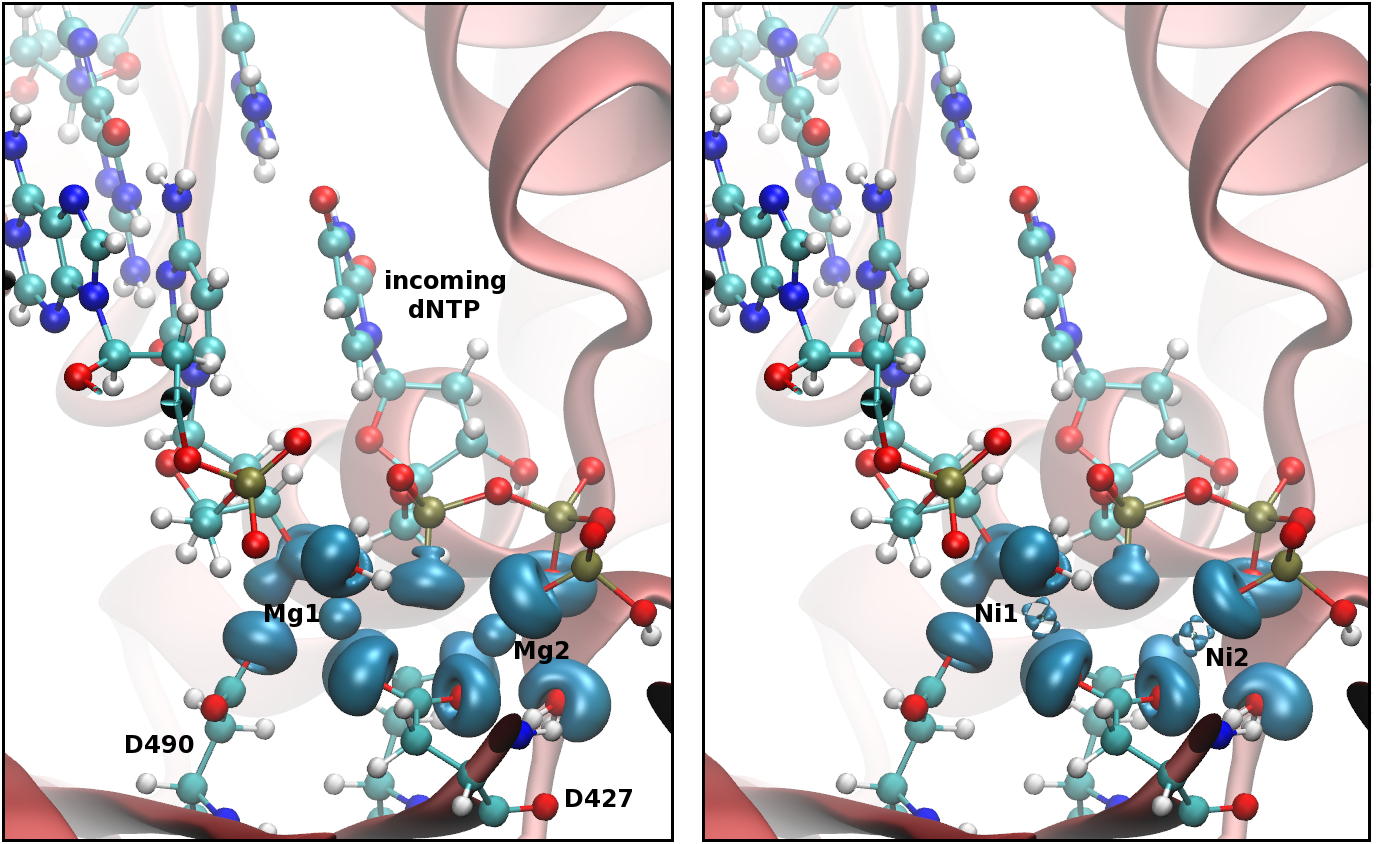
DNA alkylation is a common type of damage that can have deleterious effects. AlkB and its human homologues, ABH1-8 and FTO, catalyze a direct de-alkylation of different DNA and RNA substrates.
Computational Methods Development
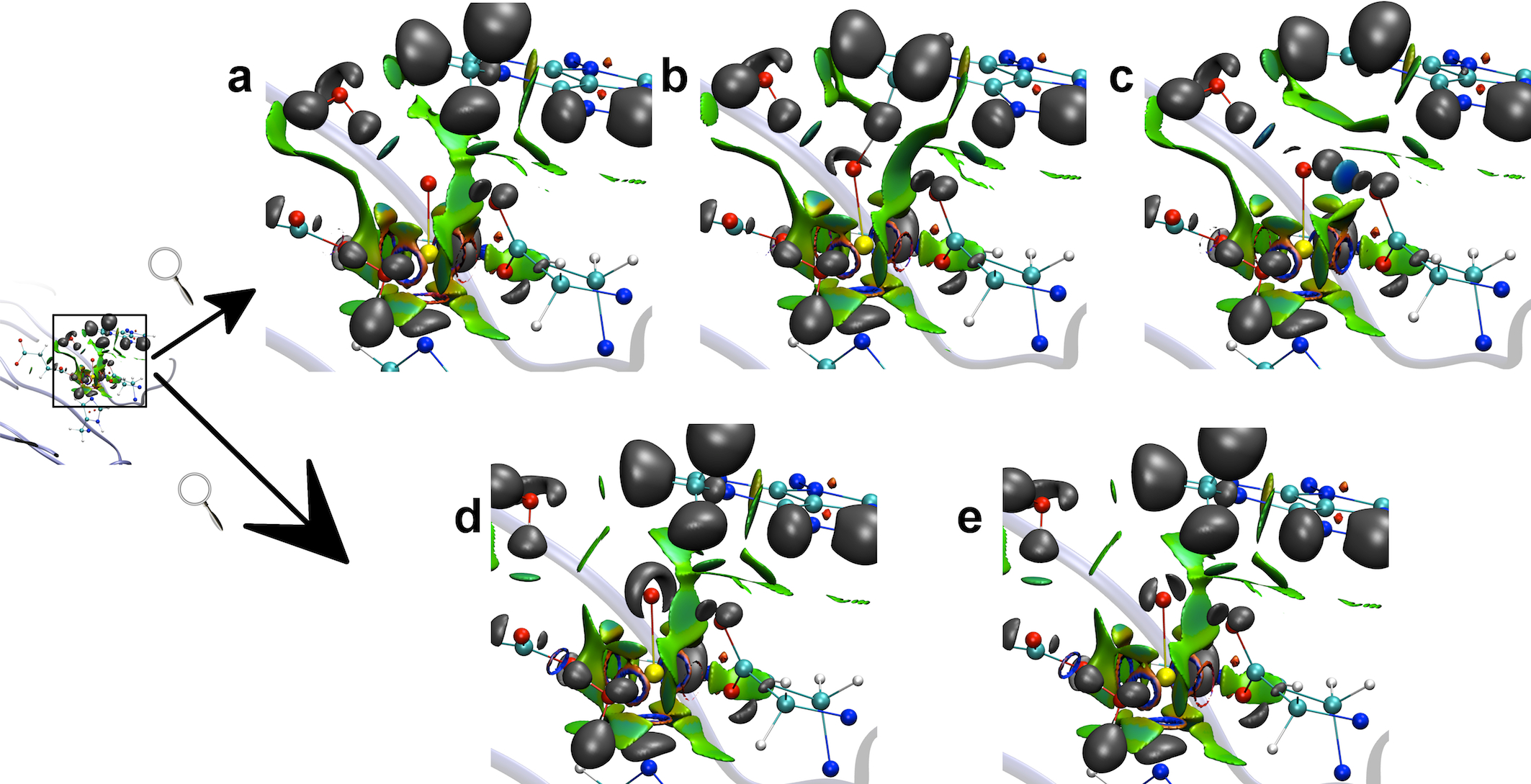
Conventional force fields (FFs) are parametric functions that calculate the energies and forces of the systems of interest. These functions use terms that reproduce bonded (bond length, angle, etc.) and non bonded (electrostatics, Van der Waals) properties using parameters obtained from experimental and theoretical determinations. In general, these FFs approximate the calculation of the electrostatic (Coulomb) interaction by using a collection of either point charges or multipoles (pairwise additive). We are involved in the development of a new FF named Gaussian Electrostatic Model (GEM). This new force field uses frozen electronic density of molecular fragments to calculate intermolecular interactions. The use of frozen densities results in higher accuracy for molecular properties and intermolecular interactions.
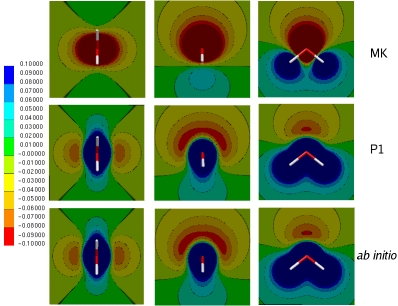
We have developed an initial model for water that combines the Coulomb and Exchange terms from GEM with the polarization, (modified) van der Waals and bonded terms from AMOEBA, we term this model GEM*:
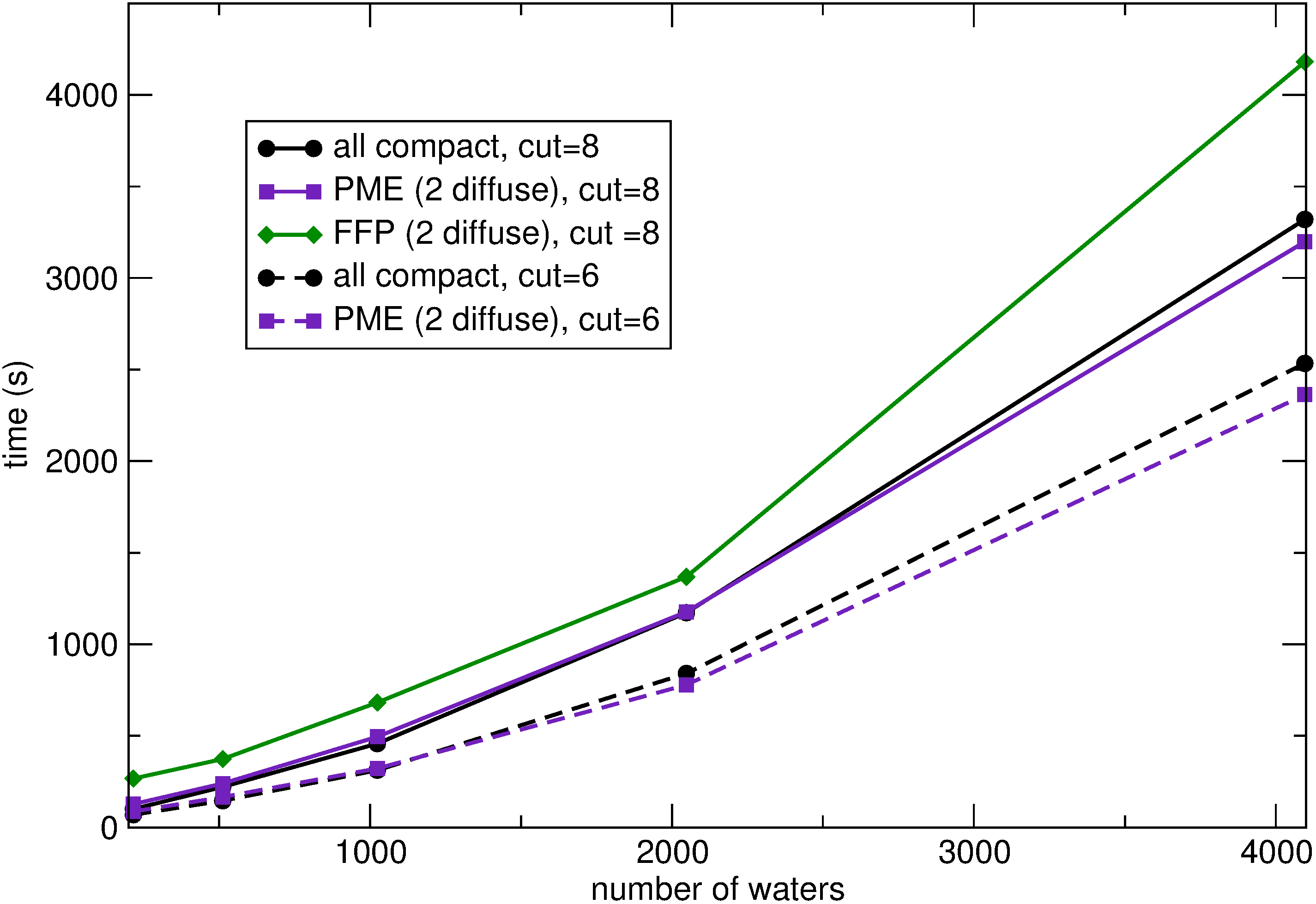
We are also working on the development of advanced force fields for Ionic Liquids. To this end, we are using the AMOEBA force field as a starting point for imidazolium-based ion pairs.
Biomarker Determination
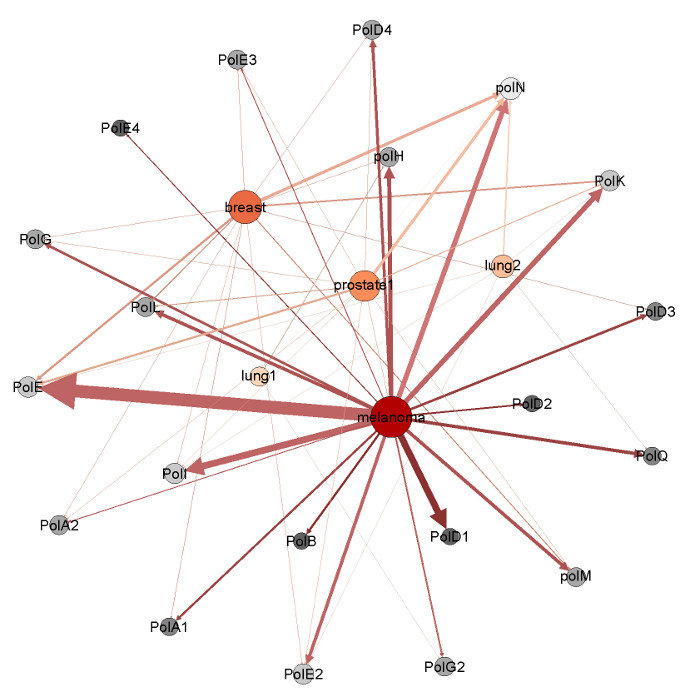
Our work on DNA repair has resulted in our interest in bioinformatics. We have developed a new method called Hypothesis Driven-SNP-Search (HyDn-SNP-S), which allows the determination of disease related SNPs for any disease (see ref. 40).
Quantum Computing
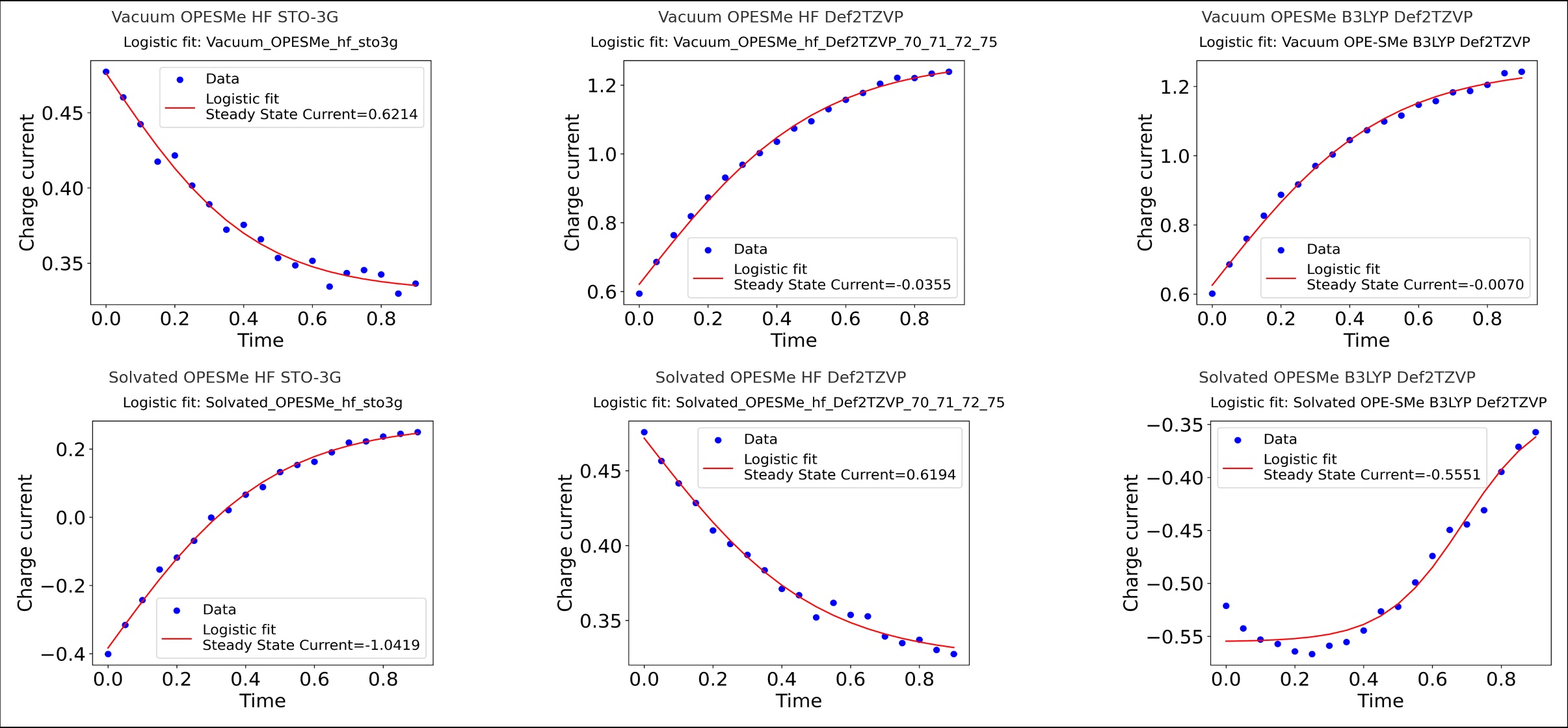
We are exploring the use of noisy intermediate-scale quantum computing devices for the simulation of electron transport in complex solvated molecular systems. We are leveraging our expertiese in polarizable QM/MM simulations to obtain initial states to generate molecular Hamiltonians for complex systems, which are employed via active space transformers for NISQ simulations. We are using this approach to investigate electron transport in Oligophenylethynylene-sulfurmethyl in vacuum and THF solution, and in doped DNA.
In addition, we are interested in the development of methods for the computational investigation of enzyme reactions. One way to perform these calculations is by combining quantum mechanics and molecular mechanics (QM/MM). QM/MM calculations allow the study of enzyme reactions by simulating the reacting part of the system at the QM level while allowing the simulation of the rest of the protein by a much faster classical method. We are interested in developing methods to improve accuracy and speed of QM/MM calculations.